Mô-đun tổng hợp TIANSeq RNA Frag / cDNA
Đặc trưng
■ Hiệu suất chuyển đổi cao: Hệ thống phản ứng được tối ưu hóa đảm bảo tính ổn định của sản phẩm và hiệu quả cao của quá trình tổng hợp cDNA sợi đôi.
■ Vận hành đơn giản: Quy trình phản ứng tích hợp và các bước vận hành đơn giản hóa.
■ Tính tương thích tốt: cDNA sợi đôi tinh khiết của sản phẩm phản ứng có thể được sử dụng trực tiếp để xây dựng thư viện RNA-Seq.
Sự chỉ rõ
Kiểu: Sự phân mảnh RNA và tổng hợp cDNA trong việc chuẩn bị thư viện RNA không định hướng
Mẫu vật: RNA tổng số
Mục tiêu: mRNA
Bắt đầu nhập mẫu: Tổng số mẫu RNA là 10 ng-1 μg và của mẫu mRNA thấp nhất là 500 pg
Thời gian hoạt động: 2,5 giờ
Các ứng dụng hạ nguồn: Sửa chữa cuối và nối đuôi 3 'cuối dA của cDNA sợi đôi
Tất cả các sản phẩm có thể được tùy chỉnh cho ODM / OEM. Để biết chi tiết,vui lòng nhấp vào Dịch vụ tùy chỉnh (ODM / OEM)




Hiện tại, công nghệ giải trình tự thông lượng cao chủ yếu dựa trên công nghệ giải trình tự thế hệ tiếp theo. Vì độ dài đọc của công nghệ giải trình tự thế hệ tiếp theo bị hạn chế, chúng ta phải chia trình tự có độ dài đầy đủ thành các thư viện phân mảnh nhỏ để sắp xếp thứ tự. Theo nhu cầu của các thí nghiệm giải trình tự khác nhau, chúng tôi thường chọn giải trình tự kết thúc đơn hoặc giải trình tự kết thúc kép. Hiện tại, các đoạn DNA của thư viện giải trình tự thế hệ tiếp theo thường phân bố trong khoảng 200-800 bp.
a) ADN kém chất lượng và chứa chất ức chế. Sử dụng các mẫu DNA chất lượng cao để tránh ức chế hoạt động của enzyme.
b) Lượng mẫu ADN không đủ khi sử dụng phương pháp không dùng PCR để xây dựng thư viện ADN. Khi đầu vào của DNA phân mảnh vượt quá 50 ng, quy trình làm việc không có PCR có thể được thực hiện một cách chọn lọc trong quá trình xây dựng thư viện. Nếu số lượng bản sao của thư viện quá thấp để được giải trình tự trực tiếp, thư viện DNA có thể được khuếch đại bằng PCR sau khi thắt nút bộ điều hợp.
c) Ô nhiễm RNA dẫn đến định lượng DNA ban đầu không chính xác Sự ô nhiễm RNA có thể tồn tại trong quá trình tinh sạch DNA bộ gen, có thể dẫn đến định lượng DNA không chính xác và tải DNA không đủ trong quá trình xây dựng thư viện. RNA có thể được loại bỏ bằng cách xử lý với RNase.
A-1
a) Xuất hiện các mảnh nhỏ (60 bp-120 bp) Các mảnh nhỏ thường là các mảnh tiếp hợp hoặc dimer do bộ tiếp hợp tạo thành. Việc làm sạch bằng hạt từ tính Agencourt AMPure XP có thể loại bỏ hiệu quả các mảnh bộ điều hợp này và đảm bảo chất lượng trình tự.
b) Các đoạn lớn xuất hiện trong thư viện sau khi khuếch đại PCR Kích thước của đoạn DNA trong thư viện sẽ tăng thêm 120 bp sau khi bộ tiếp hợp được nối. Nếu đoạn DNA tăng hơn 120 bp sau khi thắt bộ điều hợp, nó có thể là do sự khuếch đại đoạn bất thường của quá trình khuếch đại PCR. Giảm số chu kỳ PCR có thể ngăn chặn tình trạng này.
c) Kích thước bất thường của các đoạn DNA thư viện sau khi thắt bộ tiếp hợp Chiều dài của bộ tiếp hợp trong bộ dụng cụ này là 60 bp. Khi hai đầu của đoạn được nối với các bộ tiếp hợp, chiều dài sẽ chỉ tăng thêm 120 bp. Khi sử dụng bộ chuyển đổi khác với bộ chuyển đổi được cung cấp bởi bộ này, vui lòng liên hệ với nhà cung cấp để cung cấp thông tin liên quan như chiều dài bộ chuyển đổi. Hãy đảm bảo rằng quy trình làm việc và hoạt động thử nghiệm tuân theo các bước được mô tả trong sách hướng dẫn.
d) Kích thước đoạn DNA bất thường trước khi thắt bộ tiếp hợp Nguyên nhân của vấn đề này có thể do điều kiện phản ứng sai trong quá trình phân mảnh DNA. Thời gian phản ứng khác nhau nên được sử dụng cho đầu vào DNA khác nhau. Nếu đầu vào DNA lớn hơn 10 ng, chúng tôi khuyên bạn nên chọn thời gian phản ứng là 12 phút làm thời gian bắt đầu để tối ưu hóa và kích thước đoạn được tạo ra tại thời điểm này chủ yếu nằm trong khoảng 300-500 bp. Người dùng có thể tăng hoặc giảm độ dài của đoạn ADN trong 2-4 phút tùy theo yêu cầu của mình để tối ưu hóa đoạn ADN với kích thước cần thiết.
A-2
a) Thời gian phân mảnh không được tối ưu hóa Nếu DNA bị phân mảnh quá nhỏ hoặc quá lớn, vui lòng tham khảo Hướng dẫn lựa chọn thời gian phân mảnh được cung cấp trong hướng dẫn để xác định thời gian phản ứng và sử dụng mốc thời gian này làm đối chứng, đồng thời thiết lập thêm hệ thống phản ứng kéo dài hoặc rút ngắn 3 phút để điều chỉnh chính xác hơn về thời gian phân mảnh.
A-3
Sự phân bố kích thước bất thường của DNA sau khi xử lý phân mảnh
a) Phương pháp rã đông không đúng đối với thuốc thử phân mảnh hoặc thuốc thử không được trộn lẫn hoàn toàn sau khi rã đông. Làm tan băng thuốc thử 5 × Fragmentation Enzyme Mix trên đá. Sau khi rã đông, trộn đều thuốc thử bằng cách vuốt nhẹ đáy ống. Không xoáy thuốc thử!
b) Mẫu đầu vào DNA có chứa EDTA hoặc các chất ô nhiễm khác Sự cạn kiệt các ion muối và chất chelat trong bước tinh sạch DNA là đặc biệt quan trọng đối với sự thành công của thí nghiệm. Nếu DNA được hòa tan trong 1 × TE, hãy sử dụng phương pháp được cung cấp trong hướng dẫn để thực hiện phân mảnh. Nếu nồng độ EDTA trong dung dịch không chắc chắn, thì nên làm sạch DNA và hòa tan nó trong nước khử ion cho phản ứng tiếp theo.
c) Định lượng ADN ban đầu không chính xác Kích thước của ADN phân mảnh có quan hệ mật thiết với số lượng ADN đầu vào. Trước khi xử lý phân mảnh, việc định lượng chính xác DNA bằng Qubit, Picogreen và các phương pháp khác là điều cần thiết để xác định chính xác lượng DNA trong hệ phản ứng.
d) Việc chuẩn bị hệ thống phản ứng không theo hướng dẫn Việc chuẩn bị hệ thống phản ứng phân mảnh phải được thực hiện trên nước đá theo đúng hướng dẫn. Để đảm bảo hiệu quả tốt nhất, tất cả các thành phần phản ứng nên được đặt trên đá và việc chuẩn bị hệ thống phản ứng phải được thực hiện sau khi làm nguội hoàn toàn. Sau khi chuẩn bị xong, vui lòng dùng pipet hoặc dùng pipet vuốt nhẹ để trộn đều. Đừng xoáy!
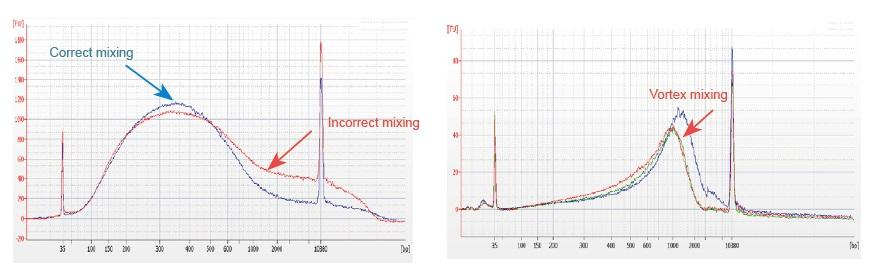
1. Trộn không đúng phương pháp (xoáy, dao động mạnh, v.v.) sẽ gây ra sự phân bố bất thường của các mảnh thư viện (như hình sau), do đó ảnh hưởng đến chất lượng của thư viện. Do đó, khi chuẩn bị dung dịch phản ứng Fragmentation Mix, vui lòng dùng pipet nhẹ lên xuống để trộn đều, hoặc dùng đầu ngón tay vuốt nhẹ cho hỗn hợp đều. Hãy cẩn thận để không trộn với xoáy.
2. DNA có độ tinh khiết cao phải được sử dụng để xây dựng thư viện
■ Tính toàn vẹn của DNA tốt: Dải điện di lớn hơn 30 kb, không có đuôi
■ OD260 / 230:> 1,5
■ OD260 / 280: 1,7-1,9
3. Lượng DNA đầu vào phải chính xác Nên sử dụng phương pháp Qubit và PicoGreen để định lượng DNA, thay vì Nanodrop.
4. Phải xác định hàm lượng EDTA trong dung dịch DNA EDTA có ảnh hưởng lớn đến phản ứng phân mảnh. Nếu hàm lượng EDTA cao, cần thực hiện tinh sạch DNA trước khi thử nghiệm tiếp theo.
5. Dung dịch phản ứng phân mảnh phải được chuẩn bị trên đá Quá trình phân mảnh nhạy cảm với nhiệt độ và thời gian phản ứng (đặc biệt là sau khi thêm chất tăng cường). Để đảm bảo độ chính xác của thời gian phản ứng, vui lòng chuẩn bị hệ thống phản ứng trên nước đá.
6. Thời gian phản ứng phân mảnh phải chính xác Thời gian phản ứng của bước phân mảnh sẽ ảnh hưởng trực tiếp đến kích thước của các sản phẩm phân mảnh, do đó ảnh hưởng đến sự phân bố kích thước của các đoạn DNA trong thư viện.
1. Loại mẫu nào được áp dụng cho bộ tài liệu này?
Loại mẫu áp dụng của bộ dụng cụ này có thể là RNA tổng số hoặc mRNA tinh khiết với tính toàn vẹn RNA tốt. Nếu RNA tổng số được sử dụng để xây dựng thư viện, bạn nên sử dụng bộ làm giảm rRNA (Cat # 4992363/4992364/4992391) để loại bỏ rRNA trước.
2. Có thể sử dụng các mẫu FFPE để xây dựng thư viện với bộ này không?
MRNA trong các mẫu FFPE sẽ bị phân hủy ở một mức độ nhất định, với tính toàn vẹn tương đối kém. Khi sử dụng bộ tài liệu này để xây dựng thư viện, nên tối ưu hóa thời gian phân mảnh (rút ngắn thời gian phân mảnh hoặc không thực hiện phân mảnh).
3. Sử dụng bước chọn kích thước được cung cấp trong sách hướng dẫn sản phẩm, điều gì có thể khiến phân đoạn được chèn có vẻ lệch nhẹ?
Việc lựa chọn kích thước phải được thực hiện theo đúng bước lựa chọn kích thước trong hướng dẫn sử dụng sản phẩm này. Nếu có sai lệch, nguyên nhân có thể là do các hạt từ tính không cân bằng với nhiệt độ phòng hoặc không được trộn đều, pipet không chính xác hoặc chất lỏng vẫn còn trong đầu hút. Nên sử dụng các đầu tip có độ hấp phụ thấp cho thí nghiệm.
4. Lựa chọn bộ điều hợp trong xây dựng thư viện
Bộ xây dựng thư viện không chứa thuốc thử bộ điều hợp và bạn nên sử dụng bộ này cùng với Bộ điều hợp chỉ mục đơn TIANSeq (Illumina) (4992641/4992642/4992378).
5. QC của thư viện
Phát hiện định lượng thư viện: Qubit và qPCR được sử dụng để xác định nồng độ khối lượng và nồng độ mol của thư viện tương ứng. Hoạt động hoàn toàn phù hợp với hướng dẫn sử dụng sản phẩm. Nồng độ của thư viện nói chung sẽ đáp ứng các yêu cầu của việc giải trình tự NGS. Phát hiện phạm vi phân phối thư viện: Sử dụng Máy phân tích sinh học Agilent 2100 để phát hiện phạm vi phân phối thư viện.
6. Lựa chọn số chu kỳ khuếch đại
Theo hướng dẫn, số chu kỳ PCR là 6-12, và số chu kỳ PCR cần thiết phải được chọn theo đầu vào mẫu. Trong các thư viện năng suất cao, sự khuếch đại quá mức thường xảy ra ở các mức độ khác nhau, được biểu hiện bằng đỉnh lớn hơn một chút sau đỉnh của dải mục tiêu khi phát hiện Máy phân tích sinh học Agilent 2100, hoặc nồng độ Qubit được phát hiện thấp hơn nồng độ của qPCR. Khuếch đại quá mức nhẹ là một hiện tượng bình thường, không ảnh hưởng đến việc giải trình tự thư viện và phân tích dữ liệu sau đó.
7. Gai xuất hiện trong hồ sơ phát hiện của Máy phân tích sinh học Agilent 2100
Sự xuất hiện của các gai khi phát hiện Máy phân tích sinh học Agilent 2100 là do các mẫu phân mảnh không đồng đều, nơi sẽ có nhiều mảnh vỡ hơn với kích thước nhất định và điều này sẽ trở nên rõ ràng hơn sau khi làm giàu PCR. Trong trường hợp này, không nên thực hiện việc lựa chọn kích thước, tức là đặt điều kiện phân mảnh ở 94 ° C trong 15 phút ủ, ở đó sự phân bố mảnh nhỏ và tập trung, và độ đồng nhất có thể được cải thiện.
Danh mục sản phẩm
TẠI SAO CHỌN CHÚNG TÔI
Kể từ khi thành lập, nhà máy của chúng tôi đã phát triển các sản phẩm đẳng cấp thế giới đầu tiên với việc tuân thủ nguyên tắc
chất lượng đầu tiên. Sản phẩm của chúng tôi đã đạt được danh tiếng xuất sắc trong ngành và có giá trị trong lòng khách hàng cũ và mới ..
- ĐT: +86 010-59822688
- Tòa nhà 5, số 86, đường Shuangying Tây, quận Trường Bình, Bắc Kinh.
- people@tiangen.com



